CPI-Predictor
Introduction
Elucidation of chemical-protein interactions (CPI) spectrum is an important topic toward understanding of the molecular polypharmacology, and plays a critical role in drug discovery. It is both time consuming and costly to determine potential CPI by experiments alone. Computationally predicting CPI network will complement researches in chemical genetics and chemogenomics.
CPI-Predictor predicts CPI spectrum based on multitarget quantitative structure-activity relationship (mt-QSAR) strategy using supporting vector machine algorithm. Two comprehensive CPI networks of 81,689 CPI binary pairs between 50,924 compounds and 136 G protein-coupled receptors (GPCRs) and 43,965 CPI binary pairs between 23,376 compounds and 176 kinase domains were collected from ChEMBL database for model buildings (Figure 1). The range of the area under the receiver operating characteristic curve (AUC) for the test sets was 0.95 to 1 and 0.82 to 1 for 100 GPCRs and 100 kinases mt-QSAR models, respectively. The server of CPI-Predictor would be much helpful to discover the molecular polypharmacology, off-targets and drug repositioning focused on G protein-coupled receptors and kinome.
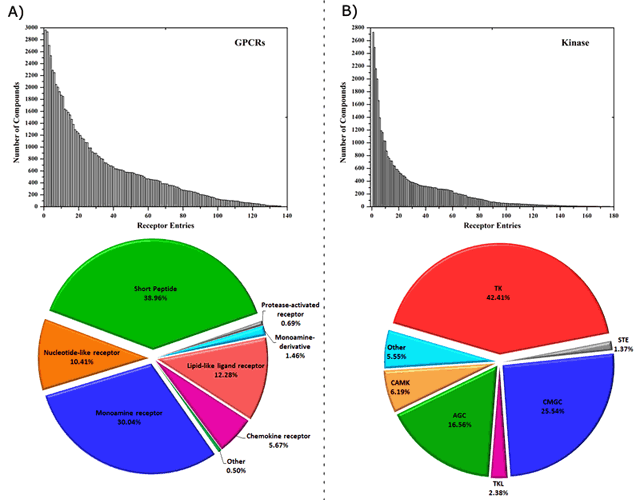
Figure 1. CPI-Predictor cover diverse ligands and target distribution.
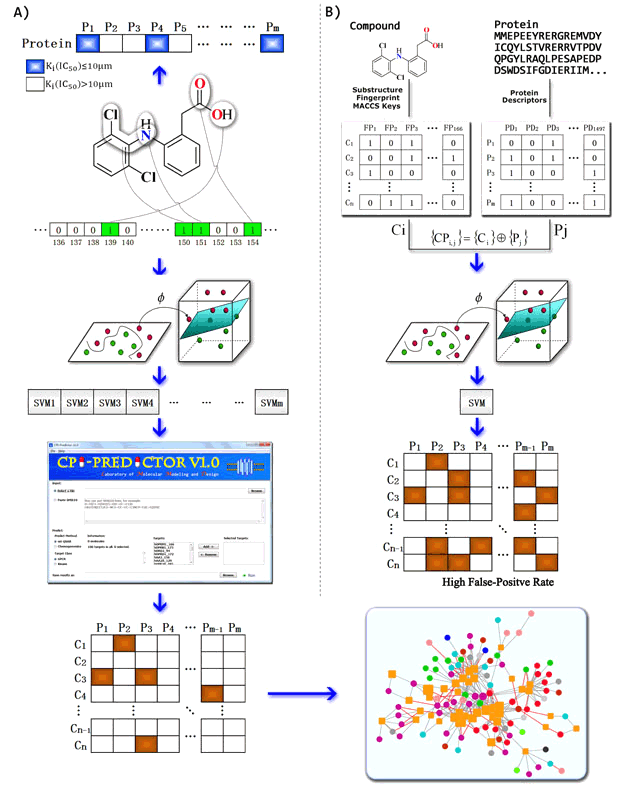
Figure 2. The workflow of multitarget quantitative structure-activity relationship (mt-QSAR) method (A) and computational chemogenomic method (B).
If you use CPI-Predictor, please cite:
1. Feixiong Cheng, Yadi Zhou, Jie Li, Weihua Li, Guixia Liu and Yun Tang*. Prediction of Chemical-Protein Interactions: Multitarget-QSAR versus Computational Chemogenomic Methods. Mol. BioSyst., 2012, 8:2373-2384
2. Jie Shen, Feixiong Cheng, You Xu, Weihua Li, and Yun Tang*. Estimation of ADME Properties with Substructure Pattern Recognition, J. Chem. Inf. Model. 2010, 50, 1034-1041.
CPI-Predictor is powered by: