NetInfer @ LMMD
|
|
NetInfer is a command-line toolkit written in C++ programming language, which can be used for predicting potential targets for approved drugs, drug candidates failed in clinical trials, and new chemical entities. It is light-weight and does not need the support of any third-party math libraries such as linear algebra libraries. To accelerate the calculation and decrease the cost of memory space, different data structures were designed for sparse and dense matrices. NetInfer provides various functions, such as link prediction, 10-fold cross validation, leave-one-cross validation, and external validation. In a uniform platform, researchers can input their in-house data and then obtain predictive lists or evaluation indicators. Several our previous methods were implemented in this toolkit, including: | |||||||
|
Download: The latest versions are available upon request at now:
Tab-delimited text files can be used as input files for our toolkit. Each line in a input file represents one linkage between two nodes A and B, containing 5 parts:
|
||||||||
References (#Co-first authors, *Corresponding authors):
|
Tutorials
1. Introduction:
The schematic diagram of balanced substructure-drug-target network-based inference (bSDTNBI):
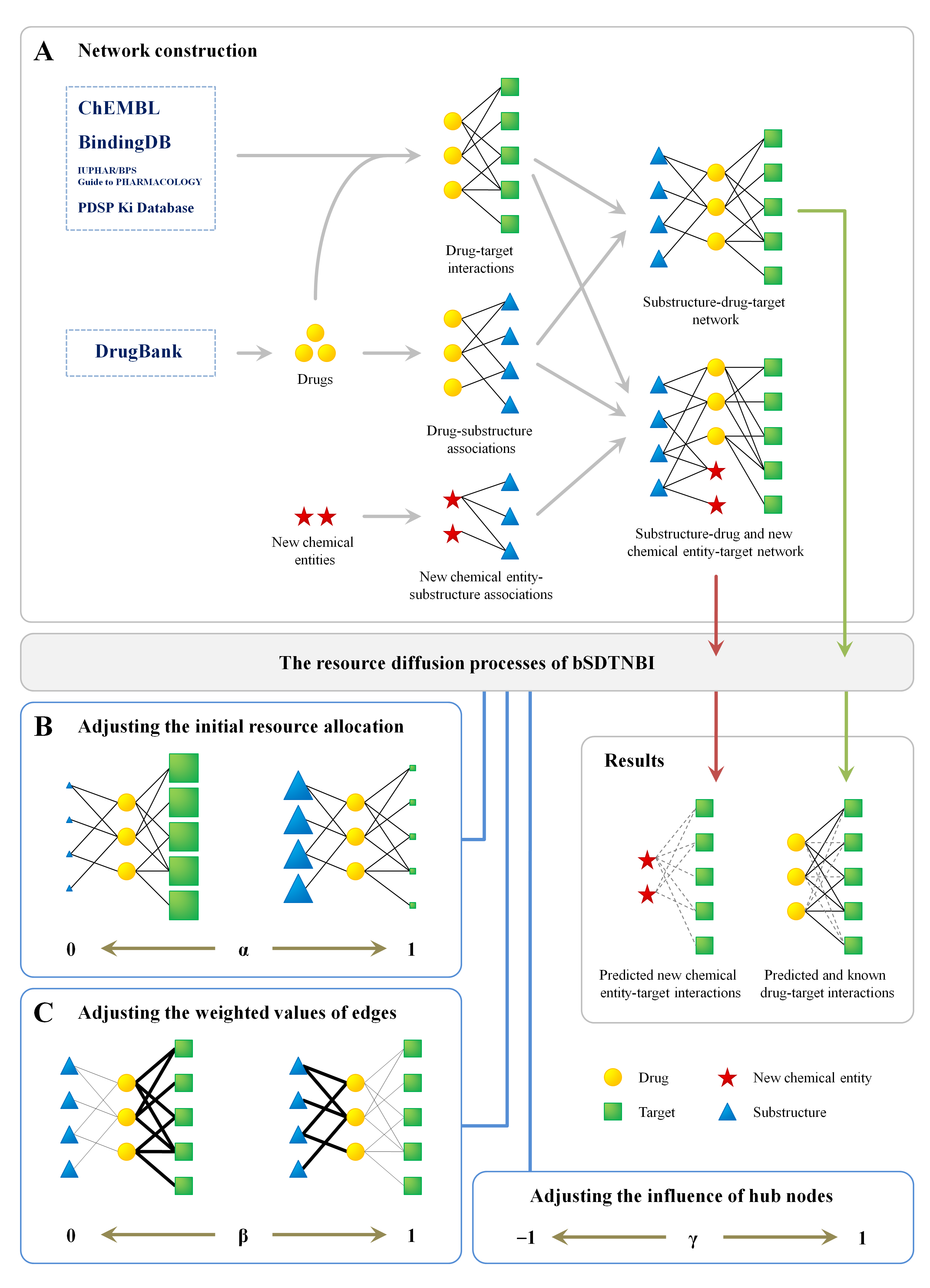
Reference:
Wu ZR#, Lu WQ#, Wu D, Luo AQ, Bian HP, Li J, Li WH, Liu GX, Huang J*, Cheng FX*, Tang Y*.
In silico prediction of chemical mechanism of action via an improved network-based inference method.
Br J Pharmacol, 2016, 173(23): 3372-3385.
DOI: 10.1111/bph.13629.
2. Global cancer drug-gene network:
We built a global cancer drug-gene network,
which provides useful information for repurposing approved therapeutic agents as novel anticancer indications,
or exploring new mechanism of action for known anticancer agents.
-
Cancer drug-gene network,
which contains:
(1) 2,100 known and 1,355 newly predicted drug-target interactions between 666 approved drugs and 369 cancer gene products,
(2) 1,533 cancer-gene associations connecting 15 cancer types/subtypes.
In this network, potential targets ranked in top 5 with highest scores for these approved drugs were predicted via the best global bSDTNBI model (Global-KR, k = 2, α = 0.1, β = 0.1, and γ = -0.5).
Reference:
Wu ZR#, Lu WQ#, Wu D, Luo AQ, Bian HP, Li J, Li WH, Liu GX, Huang J*, Cheng FX*, Tang Y*.
In silico prediction of chemical mechanism of action via an improved network-based inference method.
Br J Pharmacol, 2016, 173(23): 3372-3385.
DOI: 10.1111/bph.13629.
3. Known and predicted drug-target interactions for human GPCRs:
-
Global drug-target network for human GPCRs,
which contains known and newly predicted drug-target interactions between 25,787 known GPCR ligands and 200 human GPCRs.
In this network, potential targets ranked in top 100 with highest scores for these known GPCR ligands were predicted via the best network-based predictive model (Global-bSDTNBI-KR, k = 2, α = 0.1, β = 0.1, and γ = -0.4).
-
Local drug-target network for human GPCRs (Cytoscape format),
which only contains known drug-target interactions connecting 391 approved drugs and 113 human GPCRs.
Reference:
Wu ZR#, Lu WQ#, Yu WW, Wang TDY, Li WH, Liu GX, Zhang HK, Pang XF, Huang J, Liu MY*, Cheng FX*, Tang Y*.
Quantitative and systems pharmacology 2. In silico polypharmacology of G protein-coupled receptor ligands via network-based approaches.
Accepted.
4. Command lines:
The command lines of bSDTNBI are similar to those of SDTNBI,
except the following items:
(1) The value of parameter "-method" should be changed from "nbi" to "bnbi".
(2) Three parameters should be given in order to adjust the initial resource allocation of different node types (α),
the weighted values of different edge types (β), and the influence of hub nodes (γ), respectively.
An example of the newly added parameters (α = 0.1, β = 0.2, and γ = -0.5)
in prioritizing targets for known drugs or cross validation are:
-nbi_alpha DRUG SUB 0.1 DRUG TARGET 0.9
-nbi_beta DRUG SUB 0.2 DRUG TARGET 0.8
-nbi_gamma -0.5
An example of the newly added parameters (α = 0.1, β = 0.2, and γ = -0.5)
in predicting targets for new chemical entities or external validation are:
-nbi_alpha COMPOUND+DRUG SUB 0.1 DRUG TARGET 0.9
-nbi_beta COMPOUND+DRUG SUB 0.2 DRUG TARGET 0.8
-nbi_gamma -0.4
Page last updated at /vpmTue, 04 Aug 2026 21:53:41 +0800/222/08Asia/Shanghai310920260441/_09Asia/Shanghaipm04.p09p (Asia/Shanghai)
Copyright © 2015-2022
Laboratory of Molecular Modeling and Design,
School of Pharmacy,
East China University of Science and Technology.
All rights reserved.